
Palabras clave
Difusión en Sólidos en la nanoescala
Modelación y Simulación computacional
Cómo citar
Resumen
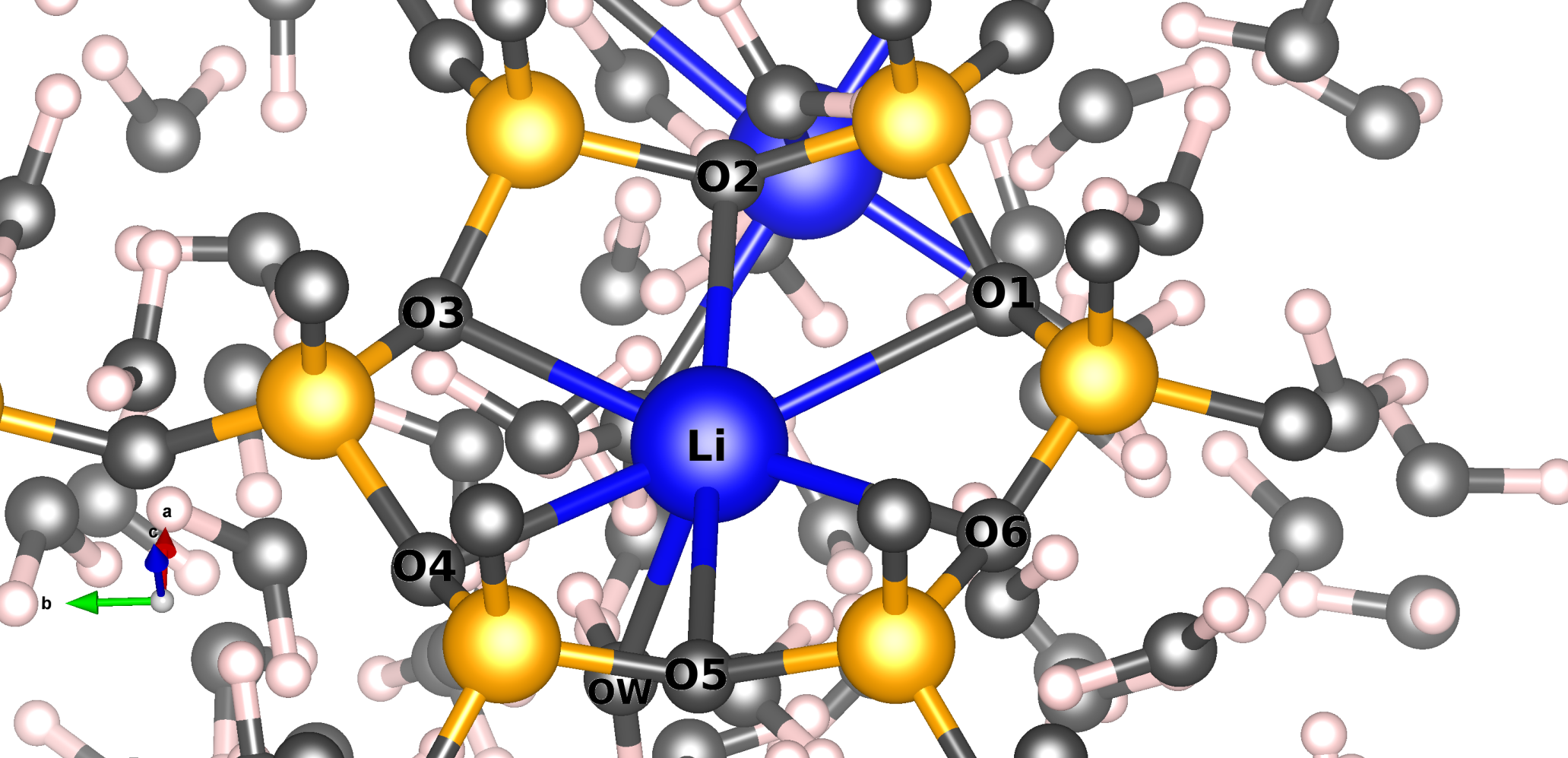
Estudiamos, usando el método de Dinámica Molecular, la movilidad de los cationes de compensación Litio en dos modelos de arcilla que se encuentran en contacto con un reservorio de agua. El sitio preferencial del Li+ es el centro del anillo hexagonal de la estructura. Las simulaciones muestran que los cationes Li+ pueden abandonar sus posiciones iniciales y desplazarse de cuatro formas diferentes. Estos movimientos son en intervalos de tiempo cortos, lo que sugiere un mecanismo de difusión por saltos. Además, nuestras simulaciones han puesto de relieve que el Li+ puede coordinar con los oxígenos del enrejado y con las moléculas de agua, y el grado de solvatación dependerá de la posición del catión. Así mismo, los cationes cercanos a la superficie son los que más difunden.

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial 4.0.
Derechos de autor 2024 Revista Cubana de Física