
Keywords
Diffusion in Nanoscale Solids
Computer modeling and Simulation
How to Cite
Abstract
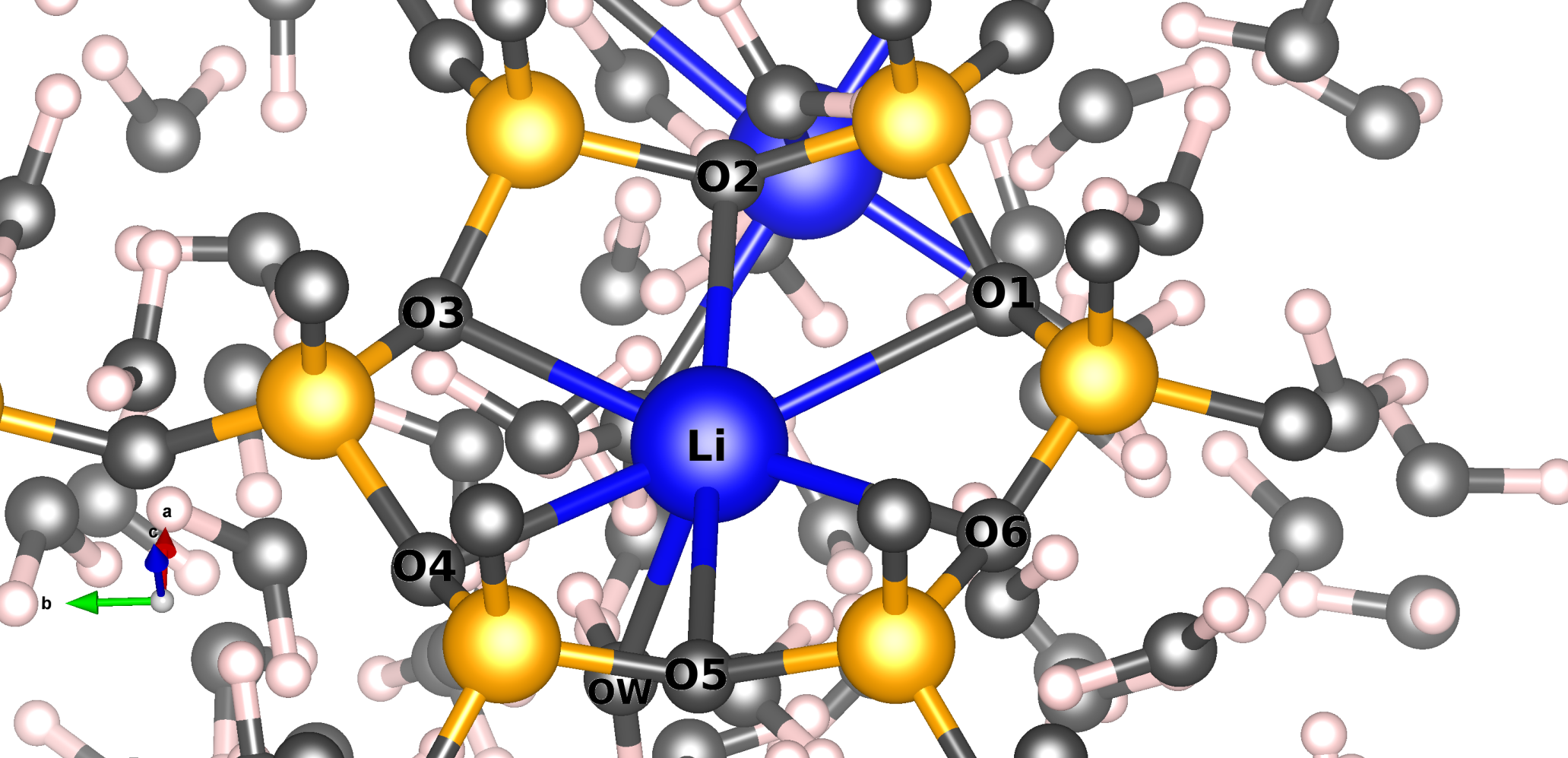
We study, using the Molecular Dynamics method, the mobility of Lithium compensation cations in two clay models that are in contact with a water reservoir. The preferential site of Li+ is the center of the hexagonal ring of the structure. The simulations show that Li+ cations can leave their initial positions and move in four different ways. These movements occur over short time intervals, suggesting a jump diffusion mechanism. Furthermore, our simulations have highlighted that Li+ can coordinate with the framework oxygens and with water molecules, and the solvation will depend on the position of the cation. Likewise, the cations close to the surface are the ones that diffuse the most.

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
Copyright (c) 2024 Sociedad Cubana de Física & Facultad de Física de la Universidad de La Habana